Quantification of parameter uncertainty
The quantification of parameter uncertainty is achieved by following a protocol [1] which aims to translate biological knowledge into informative probability distributions for every parameter, without the need for excessive mathematical and computational effort from the user. It assumes that a functioning kinetic model of the relevant biological system is already available and focuses on the procedures specific to the application of an ensemble modelling approach.
Overview of the protocol
The major protocol steps are the following:
- Collection and documentation of parameter information from the literature, databases and experiments.
- Plausibility assessment of each value collected from the literature using a standardized evidence weighting system. This includes assigning weights with regards to the quality, reliability and relevance of each experimental data point.
- Calculation of the mode (most plausible value) and Spread (multiplicative standard deviation) using the weighted parameter data sets. These values are found using a function which calculates the weighted median and weighted standard deviation of the log transformed and normalised parameter values.
- Calculation of the location (μ) and scale (σ) parameters of the log-normal distribution based on the previously defined mode and Spread.
- If required, ensure the thermodynamic consistency of the model by creating multivariate distribution for interdependent parameters (e.g., the rates of forward and reverse reactions are connected via the equilibrium constant of the reaction, and must not be sampled independently).

Procedure
Step 1: Find and document parameter values The first part of the pipeline is the collection and documentation of all available parameter values from literature, databases (e.g., BRENDA and BioNumbers) and experiments. If no values can be retrieved for a reaction occurring in a particular organism, the search can be broadened to include data from phylogenetically related organisms (the uncertainty should then be adjusted accordingly). When no values can be retrieved for a certain parameter, the search constraints must be broadened; e.g., if the Michaelis–Menten constant, KM, for a particular substrate is unknown, it might be reasonable to retrieve all KM values from a database like BRENDA in order to determine the range of generally plausible values for this parameter. In cases where no parameter values are available, an indirect estimate is often possible, based on back-of-the-envelope calculations and fundamental biophysical constraints, which will have a larger uncertainty than a direct experimental measurement but will still be informative – situations where absolutely no information about a plausible parameter range is available will be extremely rare. At this stage, it is important to collect as many experimental data with some implications for the range of plausible values as possible, including those from a diverse range of methodologies, experimental conditions and biological sources. These will be pruned and weighted in the next step. Nevertheless, in many cases, only a single experimental value will be available for a particular parameter. In the absence of any other information, it is reasonable to use this value as the mode (most plausible value) of the probability distribution. Sometimes, an experimental error (e.g., a standard deviation) is reported for the value and can be used to estimate the spread of plausible; if this is not available an arbitrary multiplicative error can be assigned, based on a general knowledge of the reliability of a particular type of experimental technology. This information, of course, can be complemented using the previous strategy of retrieving related values from databases to optimise the estimate of the spread of plausible values and avoid over-confident reliance on a single observation.

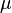 =1.5653 and
=1.5653 and 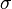 =0.4231. The mode is equal to 4 is represented by the blue line, the mean is equal to 5.2 and is represented by the green line and the median is equal to 4.8 and is represented by the yellow line. The grey area highlights the range between 1.6 and 10 within which lie 95.45% of the values, calculated by choosing a confidence interval factor of 2.5. The value 10 and the value 1.6 have equal probability of being sampled (f(4*2.5) =f(4/2.5)=0.02066).
=0.4231. The mode is equal to 4 is represented by the blue line, the mean is equal to 5.2 and is represented by the green line and the median is equal to 4.8 and is represented by the yellow line. The grey area highlights the range between 1.6 and 10 within which lie 95.45% of the values, calculated by choosing a confidence interval factor of 2.5. The value 10 and the value 1.6 have equal probability of being sampled (f(4*2.5) =f(4/2.5)=0.02066).In order to create the probability distributions, the location and scale parameters and were required. These can be easily calculated from the mean and standard deviation of the available sample data. However in many cases, there were very little or no reported values for a parameter, or there was a minimum and maximum reported value. It was therefore necessary to come up with an alternative way to derive them which at the same time would be understandable to experimentalists, without demanding complicated mathematical terms and calculations.
In order to achieve this, the mode of the log-normal distribution (global maximum) and its symmetric properties were employed. Log-normal distributions are symmetrical in the sense that values that are 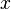 times larger than the most likely estimate, are just as plausible as values that are times smaller. More specifically, the mode of the distribution is the value
times larger than the most likely estimate, are just as plausible as values that are times smaller. More specifically, the mode of the distribution is the value 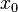 for which the condition
for which the condition 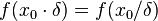 for all real numbers
for all real numbers 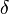 , (where
, (where 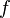 is the probability density function) is fulfilled. Hence, the user has to decide on a most plausible value for each parameter, which is set as the mode (global maximum) of the corresponding distribution (Probability Density Function or PDF), and on a range within which lie 95.45% of the values. The latter is linked to the mode via a multiplicative factor, which we call "Confidence Interval Factor". If the mode is multiplied or divided by the CI factor, the range within which 95.45% of the values are found is calculated. For instance, if the most plausible value for a parameter is
is the probability density function) is fulfilled. Hence, the user has to decide on a most plausible value for each parameter, which is set as the mode (global maximum) of the corresponding distribution (Probability Density Function or PDF), and on a range within which lie 95.45% of the values. The latter is linked to the mode via a multiplicative factor, which we call "Confidence Interval Factor". If the mode is multiplied or divided by the CI factor, the range within which 95.45% of the values are found is calculated. For instance, if the most plausible value for a parameter is 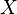 and the confidence interval multiplicative factor is
and the confidence interval multiplicative factor is 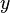 , then the mode of the distribution is set as the range where 95.45% of the plausible values are found is
, then the mode of the distribution is set as the range where 95.45% of the plausible values are found is ![[\frac{X}{y},X\cdot y]](/wiki/images/math/e/d/2/ed2bf0bf688aea7be4016cbe08252a9d.png) .
.
Based on these values, a two-by-two system of the equations containing the cumulative distribution function (CDF) and the mode is solved, in order to derive the location parameter and the scale parameter of the corresponding log-normal distribution. The equations are the following:
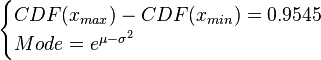
where ![CDF= \frac{1}{2}+\frac{1}{2} \mathrm{erf} \Big[\frac{lnx-\mu}{\sqrt{2}\sigma}\Big]](/wiki/images/math/e/8/a/e8ae5ac9150ce995145e521cb6f11e4e.png) and
and 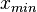 and
and 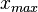 are the lower and upper bounds of the confidence interval. By substituting these into the previous equation the final form of the system is obtained:
are the lower and upper bounds of the confidence interval. By substituting these into the previous equation the final form of the system is obtained:
![\begin{cases}\frac{1}{2} \mathrm{erf} \Big[\frac{lnx_{max}-\mu}{\sqrt{2}\sigma}\Big]-\frac{1}{2} \mathrm{erf} \Big[\frac{lnx_{min}-\mu}{\sqrt{2}\sigma}\Big]=0.9545\\
Mode=e^{\mu-\sigma^{2}}\end{cases}](/wiki/images/math/9/1/1/91131156cbe90c34a6648eacf5d63c46.png)
In this way, the and parameters are obtained and from them it is easy to calculate any property in the distribution (i.e. geometric mean, variance etc.)
Parameter dependency and thermodynamic consistency
In some cases, parameters cannot be chosen separately either because they are statistically dependent, subject to thermodynamic constraints or depend on another common parameter. Therefore, thermodynamic consistency is also an important factor that needs to be considered to decide if the combinations of parameters are plausible. For instance, a very common occurrence in biological systems are forward and backward reactions. The source of dependency is the equilibrium constant, which denotes the relationship between the kinetic parameters for the "on" and "off" components of the reaction. Let’s assume a reaction that is known to have an equilibrium constant very close to 1, i.e. its standard Gibbs free energy 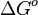 = 0. There is not much information about the rate of the reaction, so each of the two parameters is sampled from a very broad distribution. If the additional thermodynamic information is not taken into account, there will often be cases where values will be sampled from the "fast" end of the spectrum for the forward reaction rate, and from the "slow" end for the backward rate (or vice versa). Thus, inconsistent pairs of the two parameters will be generated. In this case, thermodynamic consistency requires that we discard such samples and only keep those where the two reaction rates are very similar (how similar will in turn depend on our uncertainty about the equilibrium constant).
= 0. There is not much information about the rate of the reaction, so each of the two parameters is sampled from a very broad distribution. If the additional thermodynamic information is not taken into account, there will often be cases where values will be sampled from the "fast" end of the spectrum for the forward reaction rate, and from the "slow" end for the backward rate (or vice versa). Thus, inconsistent pairs of the two parameters will be generated. In this case, thermodynamic consistency requires that we discard such samples and only keep those where the two reaction rates are very similar (how similar will in turn depend on our uncertainty about the equilibrium constant).
In order to address this problem we are employing a joint probability distribution (multivariate distribution) for the two parameters (i.e. 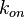 and
and 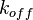 ), in order to ensure that each of the generated values for both of them are constrained within a specified range. Additionally, this approach ensures that their dependency on each other and on the equilibrium constant
), in order to ensure that each of the generated values for both of them are constrained within a specified range. Additionally, this approach ensures that their dependency on each other and on the equilibrium constant 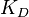 is taken into account and quantified appropriately.
is taken into account and quantified appropriately.
For instance, if the two marginal distributions are and 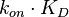 (=), is dependent on the values of and
(=), is dependent on the values of and 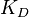 . The parameter with the largest geometric coefficient of variation (
. The parameter with the largest geometric coefficient of variation (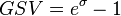 ) is usually set as the dependent one. Any product of two log-normal random variables is also log-normally distributed. Therefore, for the two log-normal distributions
) is usually set as the dependent one. Any product of two log-normal random variables is also log-normally distributed. Therefore, for the two log-normal distributions 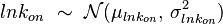 and
and 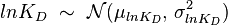 , their product will be the log-normal distribution
, their product will be the log-normal distribution 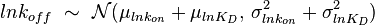 and its parameters will be
and its parameters will be 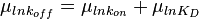 ,
, 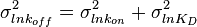 .
.
A similar strategy applies for the quotient of two log-normal distributions, although in this case the parameter will be derived by the formula 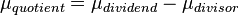 . The formula for the calculation of the parameter does not change.
. The formula for the calculation of the parameter does not change.
Thus, it becomes easy to transform the two marginal distributions and to normal ones, through the natural logarithm. The problem can therefore be reduced to the case of a multivariate normal distribution generated by the formula
Failed to parse (syntax error): f(x,y)= \frac{1}{2 \pi \sigma_X \sigma_Y \sqrt{1-\rho^2}} \exp\left( -\frac{1}{2(1-\rho^2)}\left[ \frac{(x-\mu_X)^2}{\sigma_X^2} + \frac{(y-\mu_Y)^2}{\sigma_Y^2} - \frac{2\rho(x-\mu_X)(y-\mu_Y)}{\sigma_X \sigma_Y}\right] \right)\\
where 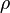 is the correlation between
is the correlation between 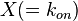 and
and 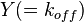 and
and 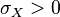 and
and 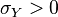 . In this case,
. In this case, 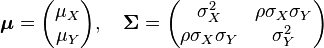 (covariance matrix).
(covariance matrix).
The parameters are assumed to be independent if = 0, so there is no correlation between them. Otherwise, the resulting bivariate iso-density loci plotted in the x,y-plane are ellipses. As the correlation parameter increases, these loci appear to be squeezed to the following line:
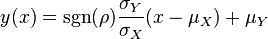
 , ) a bivariate system of distributions is created. When the two marginal distributions are non-correlated the generated points which represent parameter pairs, form a circle. When is exactly known, (the parameters and are tightly correlated through the ) the points form a straight line. Finally, if is approximately known (a distribution of values for exists) the resulting points of the bivariate system form an ellipse. The thickness and orientation of the ellipse depend on the magnitude of the correlation between the two marginal distributions and on the degree of uncertainty on the values of , and . This case represents the realistic scenario when modelling, as usually the parameter values are approximately known. The first case (no correlation) does not respect thermodynamic consistency and is therefore undesirable. The second case, although taking into account the dependency of the two parameters, is in most cases unrealistic since the value of a parameter is rarely exactly known.
, ) a bivariate system of distributions is created. When the two marginal distributions are non-correlated the generated points which represent parameter pairs, form a circle. When is exactly known, (the parameters and are tightly correlated through the ) the points form a straight line. Finally, if is approximately known (a distribution of values for exists) the resulting points of the bivariate system form an ellipse. The thickness and orientation of the ellipse depend on the magnitude of the correlation between the two marginal distributions and on the degree of uncertainty on the values of , and . This case represents the realistic scenario when modelling, as usually the parameter values are approximately known. The first case (no correlation) does not respect thermodynamic consistency and is therefore undesirable. The second case, although taking into account the dependency of the two parameters, is in most cases unrealistic since the value of a parameter is rarely exactly known.The required parameter values are obtained by generating samples from the multivariate normal distribution and then exponentiating the results.
In order to avoid errors that are introduced to the correlation matrix during the exponentiation, a matlab function called Multivariate Lognormal Simulation with Correlation (MVLOGNRAND) is used, which makes up for these errors.- ↑ Tsigkinopoulou A., Hawari A., Uttley M., Breitling R. "Defining informative priors for ensemble modelling in systems biology" (In Press)